Issue:
93
Page: 42-57
The Regulated Dietary Supplement Industry: Myths of an Unregulated Industry Dispelled
by R. William Soller, PhD, Holly J. Bayne, Christopher Shaheen
HerbalGram.
2012; American Botanical Council
Summary
In 1994, the 103rd US Congress passed the Dietary Supplement Health and Education Act (DSHEA). This legislation, along with previous laws and the 2006 Dietary Supplement and Nonprescription Drug Consumer Protection Act, has provided the US Food and Drug Administration (FDA) with statutory authority to regulate dietary supplements and those who manufacture, distribute, and sell them. It also enabled FDA to take enforcement action against unsafe or mislabeled products and those who sell them—fulfilling the agency’s mandate to protect and promote public health and safety. A comprehensive framework of regulations also has evolved through the public notice and comment rulemaking process. These regulations address essential aspects of the safety, labeling, health-related claims, and quality of dietary supplements. Consistent with its legal mandate, FDA has taken a primary role—using interagency collaborations as needed, particularly with the Federal Trade Commission (FTC)—to pursue enforcement actions against non-compliant products and companies. And yet, critics still charge that the dietary supplement industry remains “unregulated” and many members of the media use this word to characterize it. This is a myth. Today, dietary supplements represent a major industry in the national marketplace, the products of which are used widely and safely by millions of Americans under a comprehensive set of statutes of regulations.
Introduction
President Bill Clinton signed DSHEA into law on October 25, 1994.1 DSHEA amended the federal Food, Drug, and Cosmetic Act (FDCA) in several important ways, thus creating the legal framework for FDA to develop a comprehensive, predictable, and transparent regulatory system for dietary supplements. Importantly, DSHEA confirms that dietary supplements are legally classified as “food,” defines a “dietary supplement,” and clarifies that “dietary ingredients” in supplements are not “food additives” but have a distinct safety standard.* The law also requires any manufacturer or distributor of a new dietary ingredient (NDI) that is not present in the food supply prior to the date of DSHEA’s passage—and as of the date that the dietary ingredient is offered for sale—to submit to FDA a 75-day premarket notification containing safety data.† DSHEA expressly authorized FDA to prescribe Good Manufacturing Practice (GMP) regulations specific to dietary supplements. In this way, DSHEA became the central building block on which FDA crafted implementing regulations and guidance to ensure the availability of safe and properly labeled dietary supplements meeting technical standards of quality. As with other product categories under FDA’s authority, such as conventional foods, prescription and nonprescription drugs, and medical devices, the agency developed regulations and guidance specific to dietary supplements through a public rulemaking process, allowing interested parties the opportunity for input. Since the enactment of DSHEA, FDA has also issued dietary supplement-specific guidance documents addressing a range of issues, such as labeling and claims substantiation. Not unlike the development of regulations for other FDA-regulated product categories, when disagreements have arisen, as might be expected in the implementation of a new law and complex rulemakings by any government agency, stakeholders have sometimes turned to the courts to ask for judicial review of FDA’s regulatory policies. While there has been only limited litigation with respect to dietary supplements, the courts have generally upheld FDA’s statutory authority to regulate dietary supplements, much to the dismay of some in the industry.‡
Today, under DSHEA and subsequent amendments to the FDCA, FDA has broad statutory authority to regulate dietary supplements appropriately, and those who manufacture, distribute, and sell them, and to take enforcement action against unsafe or mislabeled products and those who sell them to fulfill the Agency’s mandate to protect and promote public health and safety. Nevertheless, critics still echo the accusation that dietary supplements are an unregulated industry. This is a myth.2 In fact, a comprehensive review of the available evidence strongly supports the conclusion that FDA has ample authority under current law to remove unsafe dietary supplements from the marketplace and enforce the misbranding (mislabeling) provisions of the law.3 Indeed, assessment of the agency’s enforcement activities in recent years shows that FDA is applying a risk-management approach using regulatory tools that define dietary supplements as a regulated industry. For example, as high-profile issues have arisen, FDA draws in inter-agency resources by working with groups, such as the FTC, the Drug Enforcement Administration (DEA), and the Centers for Disease Control and Prevention (CDC).
Development of the Regulatory Infrastructure for Dietary Supplements
Since the early part of the 1900s, when vitamins A and D were promoted in fish oil supplements, FDA’s predecessor agency (the Department of Agriculture, now the USDA) and what eventually became known as the dietary supplement industry clashed on a number of key issues relating to the sale and marketing of vitamins, minerals, amino acids, botanicals, and other dietary substances (now individually and collectively defined as dietary supplements).§ Their disagreements included the legality of such products, the scope of permissible health-related promotional claims, the appropriateness of marketing practices, and properly balanced federal oversight.
The major events that shaped the development of today’s comprehensive regulatory infrastructure for dietary supplements spanned over 100 years, from 1906 to 2011, culminating with the passage of the FDA Food Safety Modernization Act.4 (see Table 1). The Pure Food and Drugs Act of 1906 was enacted primarily to protect consumers from misbranded and adulterated foods and drugs moving in interstate commerce, motivated by public indignation at dishonesty and fraud in the marketplace. With its principal emphasis on foods, the 1906 Act prohibited any poisonous or deleterious substance that is injurious to health to be used in food.5 In 1938, Congress passed the FDCA, giving FDA broader authority and enforcement power over foods and drugs.** Significantly, the FDCA specifically recognized that food products may be labeled for and promoted with claims concerning effects on the structure or function of the body and for “special dietary uses.”†† FDA was also provided new authority to regulate the labeling of food for “special dietary uses.” Under section 403(j) of the FDCA, a food will be deemed misbranded “if it purports to be or is represented for special dietary uses, unless its label bears such information concerning its vitamin, mineral, and other dietary properties as the Secretary determines to be, and by regulations prescribes as, necessary in order to fully inform purchasers as to its value for such uses.”6
In the years that followed FDCA, until the passage of DSHEA in 1994, the regulatory environment for products later defined by DSHEA as dietary supplements was marked by heightened controversy, especially characterized by FDA’s attempts to restrict the sale of high-potency vitamins and minerals and limit the availability of dietary ingredients deemed not essential for human growth and nutrition. In 1976, the industry won a major victory with passage of the Proxmire Amendment, which prevented FDA from classifying vitamins and minerals as (illegal) drug products based on potency, or establishing maximum limits on the potency of vitamins and minerals in foods for special dietary use.7 Antagonism between FDA and the industry continued during the 1980s and early 1990s, particularly as the use of health claims on food labels proliferated and FDA began issuing proposed regulations implementing the Nutrition Labeling and Education Act of 1990 (NLEA).8 Industry challenged FDA in the courts and in Congress, resulting in a judicial reining in of FDA’s use of its food additive authority, and a congressionally mandated moratorium on regulations that were ultimately withdrawn. In 1994, Congress passed by unanimous consent clarifying legislation (Table 1). DSHEA emerged as viable legislation, promising both a sustainable solution to years of FDA-industry antagonism, as well as the potential to bring the stability, predictably, and transparency expected of a fully regulated industry.
FDA’s aggressive enforcement tactics ultimately forced the tipping point that created DSHEA. In the watershed decisions concerning black currant oil (made from seeds of Ribes nigrum, Grossulariaceae), then marketed as products by Traco Labs, Inc. and Oakmont Investment Co. Inc., FDA overreached by trying to regulate encapsulated black currant seed oil as a “food additive,” earning scathing criticism from the courts for the agency’s “Alice in Wonderland” approach that “defenestrates common sense.”9,10,11 There were numerous other instances of what the supplement industry and nutritional advocates considered FDA’s overreaching and strong-arm tactics.‡‡ One of America’s largest and most successful grassroots efforts ensued.12,13 An avalanche of activism overwhelmed Congress, and with a leading role played by US Senators Orrin Hatch (R-Utah) and Tom Harkin (D-Iowa), DSHEA was passed in 1994. Although not a part of official legislative history, the Senate Committee on Labor and Human Resources emphasized in its commentary “the need for Congressional action to assure citizens have continued access to dietary supplements and information about their benefits.”14
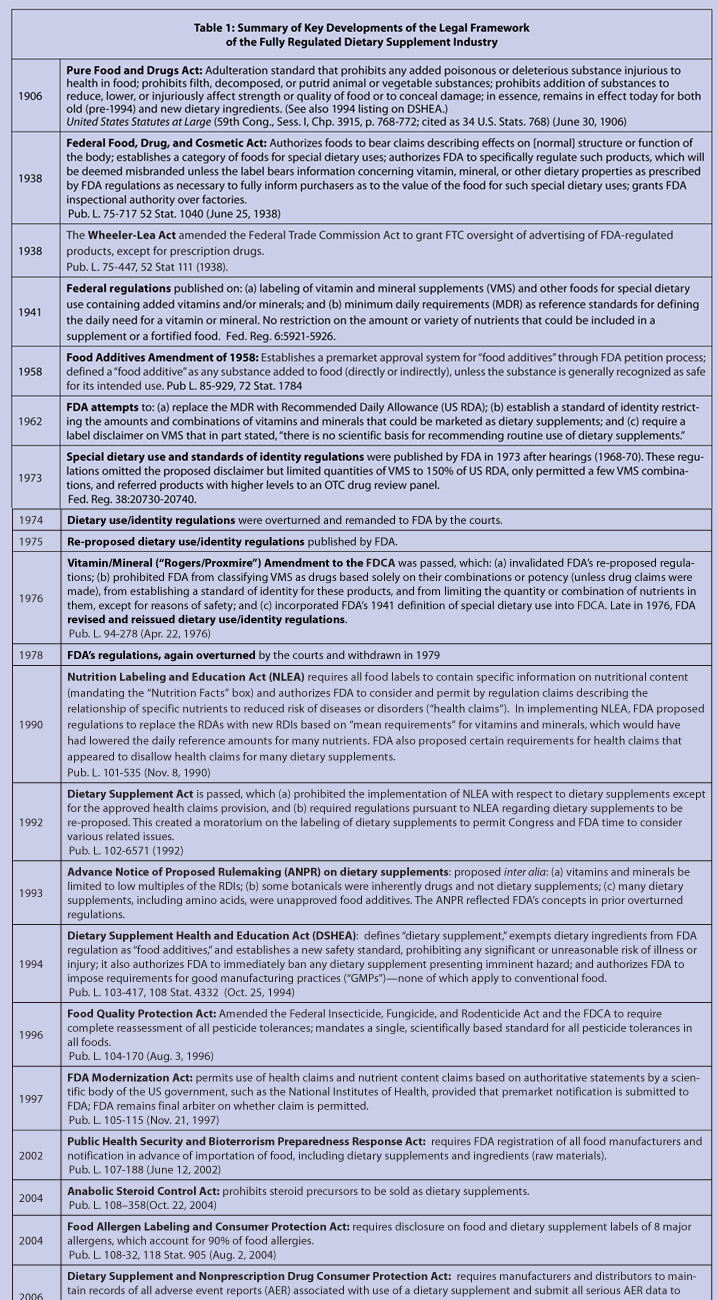
FDA Enforcement Policy and Industry Self-Policing in a Post-DSHEA Market
Criticism of dietary supplements did not dissipate after passage of DSHEA.15,16 One would think that a comprehensive law would calm critics; rather, they continued to charge that dietary supplements were an unregulated industry and that FDA had insufficient enforcement authority to protect the public from unsafe dietary supplements and unsubstantiated health-related claims.17 For its part, particularly during the immediate years following the passage of DSHEA, FDA arguably abrogated its responsibility to assertively implement DSHEA in a timely fashion. In fact, some in the dietary supplement industry have maintained that, rather than actively collaborating with the responsible majority of the industry, FDA chose a regulatory posture of studied inertia, hoping that wildcat fringe marketers would cause an implosion of the industry as a whole, resulting in a backlash against DSHEA or even its repeal.12 While there are continued rumblings that DSHEA should be amended or repealed, this has not happened, although occasional legislation proposing to amend DSHEA remains a concern for the industry. Today, there appears to be both strength of purpose from the mainstream industry and its trade associations to defend DSHEA, encourage member compliance with existing FDA regulations and guidances, and engage in more robust self-policing.§§
In the 4½ years after the passage of DSHEA, the dietary supplement industry grew exponentially to 100 million daily users and an estimated annual market of $12 billion.18 In stark contrast to this phenomenal growth in consumer acceptance of dietary supplements, what can be characterized as FDA’s non-action in the early years immediately following DSHEA limited progress in publication of comprehensive final regulations needed for a visible, predictable, and balanced regulatory process. Even so, in September of 1997, FDA published the first set of regulations implementing DSHEA, principally addressing statement of identity, ingredient and nutrition labeling of dietary supplements, nutrient content claims, procedures for notifying FDA of “statements of nutritional support” (commonly referred to as structure-function claims) in dietary supplement labeling, and procedures for filing new dietary ingredient notifications. FDA’s early period of apparent regulatory neglect began to change with a new commissioner who set the stage for policy changes. In her testimony before Congress, Jane E. Henney, MD,*** stated in 1999, “Mr. Chairman, notwithstanding our actions to date, I want to acknowledge that FDA still has a long way to go to achieve full implementation of DSHEA. I assure you that as the new Commissioner of Food and Drugs, I am focusing attention on dietary supplements, an issue that is currently a priority for FDA’s Center for Food Safety.”19 To be clear, the federal regulatory inactivity immediately post-DSHEA was not a circumstance of Dr. Henney’s doing, but rather a legacy of her predecessor, David Kessler, MD, JD,††† who remarkably devoted a mere 49 words to DSHEA in a 4,583-word address to the Food, Drug and Law Institute just 2 months after DSHEA’s passage.20
In January 2000, FDA issued a long-awaited final regulation governing dietary supplement labeling claims, establishing criteria for determining when FDA would object to a statement in dietary supplement labeling because it impermissibly suggests an effect on disease.21 This rulemaking process began in April 1998, and took almost 2 years to complete, generating significant controversy, the ire of industry, and thousands of public and health professional comments.22 Significantly, FDA had proposed to expand the regulatory definition of “disease.” Under the proposed regulation, certain natural processes such as menopause and aging, for example, would have been considered diseases, prohibiting claims in dietary supplement labeling to affect these conditions. FDA ultimately modified its course and retained the regulatory definition of “disease or health-related condition” established in the health claims rulemaking under NLEA.23 Thus, claims concerning common conditions associated with natural states or processes that would not cause significant or permanent harm if untreated are not considered as diseases under the final structure-function claims rule.
In other areas, however, FDA took a rigid and limiting approach, curtailing speech that is arguably within the scope of dietary supplement claims authorized by DSHEA (i.e., describing effects in maintaining normal, healthy bodily structure and function). For example, under the final rule, claims in dietary supplement labeling referring to supporting “healthy cholesterol levels” or the maintenance of “normal cholesterol levels” are considered by FDA as impermissible, implied disease claims, unless the claim makes it clear that cholesterol levels “are already within the normal range.” FDA’s enforcement policy in this regard represents a departure from the agency’s prior position. Under the proposed rule, a claim such as “helps maintain a healthy cholesterol level” would have been permitted in dietary supplement labeling, if truthful and not misleading. However, according to FDA in the preamble to the final regulation, “references to ‘healthy’ cholesterol may be misleading to consumers because the phrase ‘healthy cholesterol’ is now frequently used to refer to high density lipoproteins (HDL), a specific cholesterol fraction believed to be beneficial.”21
Also in January 2000, fulfilling Dr. Henney’s promise to make dietary supplements a priority, FDA’s Center for Food Safety and Applied Nutrition (CFSAN) published the Agency’s overall dietary supplement strategic plan, acknowledging that the purpose of DSHEA was to strike the right balance between providing consumers access to dietary supplements and truthful information about them, while preserving FDA’s regulatory authority to take action against supplements that presented safety problems or promoted with false or misleading labeling claims.24,25 In March of 2001, CFSAN Director Joseph A. Levitt, Esq., unveiled to Congress the agency’s report on FDA’s first comprehensive strategic plan for dietary supplements.26 The plan gave FDA’s vision for a science-based regulatory program to achieve full implementation and enforcement of DSHEA. Notably, however, real progress and finalization of key standards for dietary supplements, namely in the form of GMP regulations, were delayed. Compounded by the agency’s limited resources, tangible regulatory advancements were hampered by the necessarily lengthy process needed to obtain stakeholder and public input in developing and finalizing—through notice-and-comment rulemaking—federal standards for the manufacture of finished dietary supplements.27,28 The slowness in developing these standards was fodder for critics. Yet, high-acuity hindsight must appropriately recognize the complexities of the issues requiring a disciplined and deliberative approach to developing a rational and sustainable regulatory infrastructure for a major industry.
The Effect of the Ephedra Controversy
In February 2004, following years of contentious rulemaking that began in 1997, FDA prevailed in issuing a final rule declaring dietary supplements containing ephedrine alkaloids (mainly from ephedra [Ephedra sinica, Ephedraceae]) to be illegal (adulterated) under DSHEA’s “significant or unreasonable” risk safety standard.29 Ephedra-containing dietary supplements—particularly those based on relatively high concentrations of its extracts—promoted for weight loss and athletic performance, rose in popularity during the immediate post-DSHEA years and were characterized as being associated with cardiovascular events and other adverse product experiences. In issuing the regulation, FDA applied a risk/benefit standard, determining that dietary supplements containing ephedrine alkaloids are adulterated under the FDCA because they present an unreasonable risk of illness or injury under the conditions of use recommended in labeling or under ordinary conditions of use, without conferring any benefit.‡‡‡
A legal challenge to the final regulation ensued, with plaintiffs challenging FDA’s use of risk/benefit analysis to ban dietary supplements containing low levels of ephedrine alkaloids without any scientific proof of a significant or unreasonable risk of illness or injury at such levels. Although plaintiffs prevailed at the district court level, the Court of Appeals reversed the lower court’s decision, essentially upholding FDA’s decision to ban dietary supplements containing ephedra, regardless of the level of concentration or directions in product labeling. The court also found that FDA was not arbitrary and capricious in its Final Rule banning ephedrine alkaloids and ephedra, and that FDA had met its evidentiary burden to justify a total ban. The petitioner’s subsequent efforts for re-hearing by the Court of Appeals and review by the US Supreme Court were unsuccessful. Notably, the ephedra situation highlighted a safety-related and post-marketing surveillance gap that had yet to be filled by FDA—mandatory reporting of serious adverse product experiences associated with dietary supplements.
In December 2006, the Dietary Supplement and Nonprescription Drug Consumer Protection Act (DSNDCPA) was signed into law, requiring companies engaged in the sale and distribution of dietary supplements and non-prescription over-the-counter (OTC) drugs to maintain records of all adverse event reports received by a company, to report all serious adverse events to FDA, and to make such reports available to the agency on request. Several years earlier, the American Herbal Products Association (AHPA), a national trade association consisting of herbal and botanical product companies, filed a Citizen Petition with FDA (March 2003), recommending that the agency impose a mandatory reporting requirement on dietary supplement companies for all “serious adverse event” reports associated with use of a dietary supplement. AHPA’s petition requested that FDA establish such requirements through notice and comment rulemaking, modeled on requirements for prescription drug products. The DSNDCPA was not opposed by responsible mainstream components of the dietary supplement and OTC drug product industries.
The Senate Report accompanying the bill, introduced in June 2006 by Senator Hatch, stated the legislation “will enhance the Agency’s effort to identify potential public health issues associated with the use of these products” and “will give consumers greater assurance that public health officials are on top of emerging, serious safety problems so they can take immediate, appropriate action.”30 The law requires the company whose name appears on the label of a dietary supplement (manufacturer, packer, or distributor) to maintain records of all adverse event reports associated with use of a dietary supplement for a period of 6 years. Reports of “serious adverse events” must be filed with FDA using the mandatory “MedWatch” Form 3500A (the same form used for prescription and OTC drugs and medical devices) within 15 days of their receipt. A “serious adverse event” is defined as an event that results in death, a life-threatening experience, inpatient hospitalization, a persistent or significant disability or incapacity, or a congenital anomaly or birth defect associated with the use of a dietary supplement. Additionally, a serious adverse event includes medical or surgical intervention, based on reasonable medical judgment that is necessary to prevent any of the outcomes listed above.§§§ Records of all adverse event reports must be made available to FDA upon request, and may be inspected by FDA during a routine GMP inspection.31 In October 2007, FDA issued draft guidance for industry regarding dietary supplement adverse event reporting and recordkeeping requirements, followed by revised guidance in June 2009, although comments to the guidance may be submitted at any time.32 The guidance describes the minimum data elements for filing serious adverse event reports to FDA, clarifies the criteria used to determine whether an adverse event report would be considered serious by FDA, and addresses a company’s recordkeeping obligations.
FDA’s implementation of the dietary supplement provisions of the DSNDCPA is consistent with the agency’s approach to monitoring the safety of drugs, medical devices, and other FDA-regulated products. Dietary supplement adverse event and serious adverse event reports filed by consumers, health professionals, and industry are maintained by FDA in the CFSAN Adverse Event Reporting System (CAERS), along with adverse event data on foods, medical foods, infant formulas, color additives, and cosmetics. Data are entered by FDA using “MedDRA” medical reporting terminology, the same terminology used to capture adverse event data on drugs, biologics, vaccines, and medical devices.33 Every entry into the CAERS database is reviewed by an FDA medical officer to evaluate the strength of the evidence suggesting that the dietary supplement caused the adverse event, using categories (e.g., certain, probable, unlikely) described in the World Health Organization’s (WHO’s) publication Safety Monitoring of Medicinal Products: Guidelines for Setting Up and Running a Pharmacovigilence Center (2000).34 FDA’s review process is designed to determine whether a “signal” has been generated by a number of reports (a cluster) in the database suggesting a particular product or ingredient may present a safety problem. If a signal has been identified suggesting a safety problem associated with a dietary supplement, FDA will inspect the manufacturer (or distributor) and inspect the company’s adverse event reports to collect additional information about the product. FDA will also convene a “Health Hazard Evaluation Board” consisting of CFSAN’s Chief Medical Officer and other FDA medical officers to evaluate all of the data and prepare a written report.35 FDA effectively used this process in the case of the multi-ingredient dietary supplement “Hydroxycut,” which was associated with 23 liver-related adverse effects (idiosyncratic hepatotoxicity) between 2002 and 2009.36 At FDA’s request, Iovate Health Sciences, Inc., initiated a massive recall of Hydroxycut in the United States and Canada, recalling over 21 million units of product. In considering a range of enforcement options available to the Agency, FDA will also conduct a risk-benefit analysis of the product under section 402 (f) of the FDCA to determine whether the dietary supplement presents a significant or unreasonable risk to public health or presents an imminent hazard. FDA’s enforcement policy is that dietary supplements generally do not provide significant benefits to outweigh any public health risk and allow for continued marketing, unlike prescription drug products for which greater risk is tolerated based on benefits of treating disease.
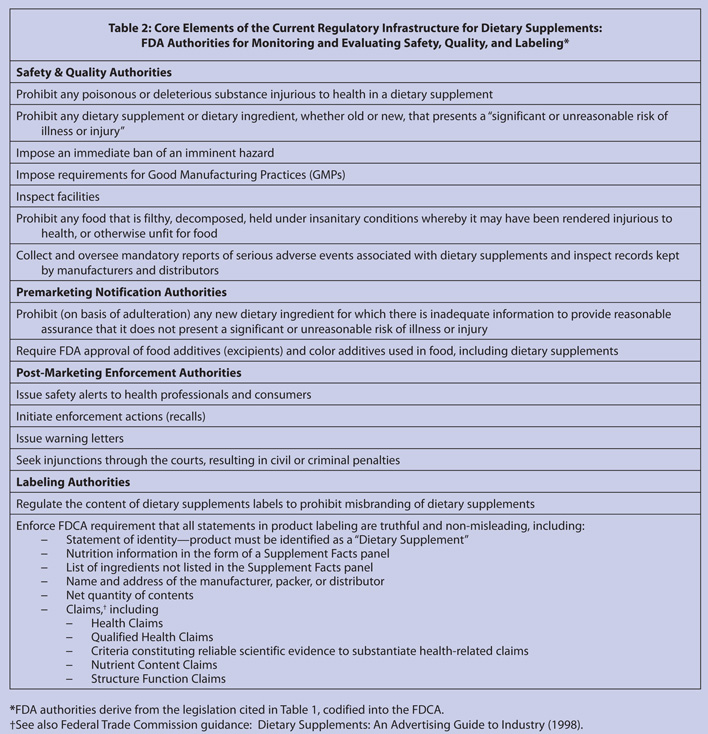

Regulation of Current Good Manufacturing Practices
In June 2007, 13 years following DSHEA’s enactment, FDA issued final regulations governing dietary supplement GMPs. DSHEA authorized FDA to prescribe GMPs for dietary supplements through notice and comment rulemaking.37 This was particularly relevant since it is generally accepted by experts and responsible segments of the industry that FDA’s food manufacturing, packaging, and hygiene standards that establish GMPs for conventional foods are inadequate for ensuring dietary supplement quality, ingredient identity, and potency as claimed in product labeling. DSHEA required FDA to model dietary supplement GMPs on food GMPs. FDA’s progress in issuing the 2007 final regulation was agonizingly slow, spanning about 10 years. Industry proposed a GMP standard to FDA in the fall of 1995, which was essentially published by FDA as an Advanced Notice of Proposed Rulemaking in February 1997. Following industry outreach efforts, FDA published a proposed rule in 2004.38,39 The final rule in 2007 generally reflects industry’s proposed standards, in that they were designed to ensure consumers are provided with dietary supplements that: (1) are safe and not adulterated or misbranded; (2) have the identity and provide the quantity of dietary ingredients declared in labeling; and (3) meet the quality specifications that the supplement is represented to meet.
The dietary supplement GMP regulations mandate that manufacturers of dietary supplements establish a comprehensive system of controls and strictly document each stage of the manufacturing process to help ensure that products have the identity, purity, strength, and composition stated on product labels and that finished dietary supplements meet clearly defined specifications in the manufacturing record and are not adulterated. The final rule establishes the minimum current GMP necessary for activities relating to the manufacturing, packaging, labeling, and holding of dietary supplements to ensure product quality.**** Failure to follow dietary supplement GMP is deemed to adulterate a product under the FDCA, which could subject the product to FDA enforcement action.40
Issuance of the final dietary supplement GMP regulations marked a major regulatory achievement for FDA and significantly raised the bar of compliance for dietary supplement manufacturers. Notably, the regulation allowed for a 3-year staggered compliance period, with large companies (500 or more full-time employees) becoming subject to the rule in June 2008, followed by medium companies the following year and small companies (fewer than 20 employees) in June of 2010. FDA has made enforcement of the dietary supplement GMPs a priority. FDA GMP inspections of dietary supplement facilities have risen progressively from 12 in 2008 to 75 in 2010.41 Of particular note is what appears in one of FDA’s first warning letters relating to enforcement of the current GMP regulations for dietary supplements.42 Table 3 shows a list of FDA’s major findings in a recent inspection of a dietary supplement firm, demonstrating the robustness of the GMP regulations in mandating strict quality control procedures from start to finish, to ensure the quality of raw materials and finished products.
Both FDA and industry are supported by non-governmental organizations in meeting their respective twin goals: FDA to ensure that dietary supplements are safe and not adulterated under the law; and industry to ensure compliance and that only properly labeled dietary supplements meeting technical standards of quality reach consumers. The United States Pharmacopeial Convention (USP) is a nonprofit scientific organization that has been setting standards for food and drug ingredients since 1820 as part of its mission to improve the quality and safety of foods and drugs moving in international commerce. Compendial standards established by USP have legal effect under the FDCA and are enforceable by FDA. Under the DSHEA amendments to the FDCA, a dietary supplement will be deemed misbranded if it is falsely represented as conforming to the specifications of an official compendium (such as USP monographs).43
In 1995, USP members adopted a resolution to establish standards for botanical and other dietary ingredients (as defined in DSHEA). Since that time, USP has published many botanical, vitamin, mineral, and other dietary ingredient quality monographs. To qualify for development of a monograph, the dietary ingredient must pass a USP safety review, according to defined criteria. USP also develops and publishes both general and specific standards, tests, assays, and other specifications for use in raw ingredient quality control operations and the manufacture of finished dietary supplements. In 2009, USP published the first edition of the USP Dietary Supplements Compendium (the “USP Compendium”), a resource of public standards and technical information for dietary supplement manufacturers that includes information developed by the major trade associations and other organizations. For example, the USP Compendium includes materials published by AHPA concerning the use of marker compounds for botanical identification and detection of adulterants and a joint AHPA-American Herbal Pharmacopoeia® (AHP) draft document concerning Good Agricultural and Collection Practices for herbal raw materials.†††† The Compendium also includes the “Standardized Information on Dietary Ingredients (SIDI™) Protocol,” developed by member companies of national trade associations, including: AHPA, the Council for Responsible Nutrition (CRN), the Natural Products Association (NPA), and the Consumer HealthCare Products Association (CHPA). The SIDI™ Protocol is designed to assist dietary ingredient suppliers in using standardized content and format templates to convey quality-related information about ingredients to downstream manufacturers of finished dietary supplements.
Analytical Methods for Ensuring Dietary Supplement Purity and Quality
In 2001, the USP Dietary Supplement Verification Program (DSVP) was launched. The program was designed to assist dietary supplement manufacturers in ensuring compliance with FDA GMP regulations. Under the program, manufacturers that demonstrate adherence to quality control procedures and undergo an audit may include a USP quality designation mark on dietary supplement labeling. To qualify for use of the USP-DSV mark, the dietary supplement must pass laboratory testing to confirm dietary ingredient identity, potency and purity, and batch-to-batch consistency.
FDA and the industry are also supported by the work of AOAC International, a nonprofit organization founded in 1884, dedicated to the development of analytical methods.44 AOAC’s official analytical methods, published in “Official Methods of Analysis of the Association of Analytical Chemists,” are incorporated by reference into FDA regulations, and are used by FDA for compliance and enforcement purposes.45 FDA’s policy is to utilize the AOAC methods of analysis in its enforcement programs when available and applicable, unless another method is prescribed by regulation. The actual work of developing and testing analytical methods for dietary supplements necessary for confirming ingredient identity, purity, and other quality parameters is conducted by AOAC’s government and industry stakeholders, with AOAC coordinating the scientific studies, receiving and evaluating the results, sanctioning acceptable and validated methods, and publishing and distributing the methods and performance data. AOAC maintains a committee structure to engage in the prioritization of methods development, based on ingredient market share and public health needs, among other factors. In 2004, under a contract with FDA, AOAC was charged with making recommendations on Best Practices for the validation of Microbiological Methods.
The Office of Dietary Supplements (ODS) within the National Institutes of Health, and the National Institute of Standards and Technology (NIST) within the US Department of Commerce are key governmental stakeholders actively involved in the development of analytical methods for dietary supplements. ODS was created in 1995, as authorized by DSHEA. The purpose and responsibilities of ODS include promoting the scientific study of dietary supplements and serving as a principal advisor to FDA on issues relating to dietary supplements. In 2002, ODS launched its Dietary Supplements Analytical Methods and Reference Materials (AMRM) program, designed to stimulate the development of validated analytical methods and reference materials to support the needs of industry, regulators (namely FDA), contract laboratories, and researchers. The goal of the AMRM program is to develop and validate methods that can be used to reliably identify and quantify ingredients in dietary supplements, including biologically active compounds, markers, and contaminants and other adulterants. To achieve this goal, ODS collaborates with FDA, AOAC, USDA, and NIST, among other organizations.
NIST is a non-regulatory federal agency whose mission is to promote domestic innovation and industrial competitiveness by advancing science, standards, and technology to enhance national security and improve quality of life. While the work of NIST is not widely known, it is nonetheless is making a significant contribution to dietary supplement quality control efforts, particularly in the area of development of botanical Standard Reference Materials (SRMs), which are needed to support adherence to current GMPs and ensure product quality. For example, NIST has developed 2 natural-matrix SRMs for saw palmetto (Serenoa repens, Arecaceae) fruit.46
Other non-governmental organizations also are engaged in assisting dietary supplement manufacturers and ingredient suppliers in complying with FDA’s GMP regulations and ensuring product quality. NSF International—an independent nonprofit organization founded in 1944 to standardize food sanitation and safety requirements, develop public health standards, and certify products—administers its third-party certification program for dietary supplements that includes product testing, GMP audit inspections, and—for products that meet NSF’s criteria—use of the NSF mark in product labeling and advertising.47 The NPA, the nation’s oldest natural products trade association, primarily consisting of retailers, has instituted a GMP Certification Program using third-party auditors and designed to ensure that products supplied to member companies are produced in accordance with current GMP requirements.48
FDA Has Demonstrated DSHEA Works
Public reports from FDA’s then Associate Commissioner John Taylor, CFSAN Director Robert Brackett, and then Principal Deputy Commissioner Joshua Sharfstein in 2003, 2004, and 2010, respectively, presented the agency’s enforcement report card for dietary supplements, which documented the following actions: successful voluntary destruction of inventories, removal of websites, voluntary recalls, warning letters, cyber letters, consumer alerts, seizures and injunctions, and criminal enforcement proceedings.49,50,51 These activities show a leveraging of interagency expertise and resources. Some FDA enforcement actions are conducted in collaboration with FTC, a key federal law enforcement agency charged with regulating the advertising of dietary supplements, whose efforts are detailed in the below section on FTC.
FDA’s enforcement reports provide evidence of how FDA has exercised enforcement authority to protect public health and safety as envisioned by the agency’s strategic plan in 2000. For example, FDA has issued a number of alerts and letters to health professionals on safety issues pertaining to dietary supplement ingredients and/or products, including the following:52
Aristolochic acid: alert on liver toxicity, 2000; as found in Aristolochia spp., Asarum spp., Bragantia spp., Stephania spp., Clematis spp., Akebia spp., Cocculus spp., Diploclisia spp., Menispernum spp., Sinomenium spp., Mu tong, Fang ji, Guang fang ji, Fang chi, Kan-Mokutsu (Japanese), and Mokutsu (Japanese);
St. John’s wort (Hypericum perforatum, Clusiaceae): warning about interaction with Indinavir, 2000;
Tiractricol (or triodothryacetic acid): warning that the product is a thyroid hormone, 2000;
Comfrey (Symphytum spp., Boraginaceae): removal from market, 2001;
Dietary supplements claiming to prevent or treat Anthrax poisoning, 2001;
LipoKinetix: warning about liver injury, 2001;
Kava (or kava kava, Piper methysticum, Piperaceae): alert about liver failure, 2002;
PC-SPES® and SPES®: a multi-herbal combination adulterated with prescription drugs, 2002;
Certain Chinese diet pills: warning regarding unauthorized ingredient, fenfluramine, 2002;
ephedrine alkaloids: removal from market, 2004;
“Better than formula Ultra Infant Immune Booster 117:” warning not to buy, 2004;
Liqiang 4: warning regrading unauthorized drug, glyburide, 2005;
Red yeast rice (Monascus purpureus, Monascaceae): warning regarding unauthorized drug, lovastatin, 2007;
Colloidal silver: alert on skin and mucous membrane discoloration, 2009.
In addition, from 2001 to 2010, FDA issued a total of 314 warning letters relating to dietary supplements, including principally those relating to unapproved new drugs, adulteration, and misbranding. The relative percentage of total warning letters seems appropriate to the size of the dietary supplement industry, which is about 1% by sales (2009) of a total $2 trillion in sales represented by all FDA-regulated products for human consumption and other uses (i.e., 1% dietary supplements and herbals, 1% nonprescription medicines, 7% medical devices, 14% prescription medicines, and 77% conventional foods).53,54,55,56
FDA’s dietary supplement enforcement programs and industry outreach efforts during the past 2 years are attributable to the leadership of Margaret A. Hamburg, MD, who became the 21st Commissioner of Food and Drugs on May 18, 2009, and Joshua M. Sharfstein, MD, principal deputy commissioner under Commissioner Hamburg. Commissioner Hamburg has been charged with FDA’s implementation of President Obama’s directive for increased transparency and openness in government and in June 2009, launched FDA’s Transparency Initiative.57 A cornerstone of the Initiative is the agency’s pledge to more clearly communicate its enforcement policies and requirements to the regulated industries in order to enhance compliance. Indeed, communication to and alignment of non-government stakeholders is an important element for government agencies to use in addressing potentially serious public health issues. In December 2010, Commissioner Hamburg worked with dietary supplement trade organizations to remind companies of their legal obligations and responsibility to prevent tainted products from reaching the US market. As pointed out by the Commissioner in her open letter to the industry, products labeled as dietary supplements that contain “the same active ingredients as FDA-approved drugs, analogs of the active ingredients in FDA-approved drugs, or other compounds, such as novel synthetic steroids … do not qualify as dietary ingredients.” These undeclared ingredients may pose a potential of significant risk to consumers due to possible serious side effects and/or food-drug interactions. The letter was prompted by FDA surveillance of the dietary supplement marketplace through laboratory tests that revealed the following drug ingredients as undeclared constituents of products labeled as dietary supplements: anticoagulants (e.g., warfarin), anticonvulsants (e.g., phenytoin), HMG-CoA reductase inhibitors (e.g., lovastatin), phosphodiesterase type 5 inhibitors (e.g., sildenafil), nonsteroidal anti-inflammatory drugs (NSAIDs; e.g., indomethacin), and beta blockers (e.g., propranolol). In addition to recounting how FDA has addressed this problem through warning letters, recalls, and product seizures, the Commissioner announced fair warning that “responsible individuals and companies should be aware that the government may initiate criminal investigations to hold accountable those who violate the Federal Food, Drug, and Cosmetic Act (the Act) and endanger the public health.”58 Five major industry trade associations—CRN, NPA, United Natural Products Alliance (UNPA), CHPA, and AHPA—joined with FDA in calling for media attention to this issue and have agreed to share the letter widely within the dietary supplement industry.
Industry is on notice that, under Commissioner Hamburg, FDA is prepared to initiate criminal misdemeanor prosecutions and felony prosecutions (with a prior offense) working with the Office of Criminal Investigations (OCI), through a rejuvenation of the “Park Doctrine.” The Park Doctrine refers to a 1975 US Supreme Court case holding that a corporate official may be prosecuted for introducing misbranded or adulterated foods into interstate commerce, without proof that the official acted with intent or negligence, and even without actual awareness or knowledge of the wrongdoing.59 The requirements on corporate officials to ensure compliance with the FDCA are “onerous” and “stringent,” but as well stated by the court: “They are no more stringent than the public has a right to expect of those who voluntarily assume positions of authority in business enterprises whose services and products affect the health and well-being of the public that supports them.”60
In these examples and others not addressed here, FDA has demonstrated that under DSHEA and subsequent legislation and FDA’s currently implementing regulations, the agency is able to and does address evolving safety, quality, and labeling issues to protect the public from adulterated or mislabeled dietary supplements. Of course, FDA must be adequately supported by Congress with the level of appropriations needed for the agency to achieve its mission.
Federal Trade Commission’s Role in Regulating Dietary Supplement Advertising
FDA is supported in its mission to protect the public from false and misleading dietary supplement claims by FTC, which has broad authority to regulate dietary supplement advertising. With overlapping jurisdiction to regulate the labeling and advertising of foods, OTC drugs, medical devices, and cosmetics, FDA and FTC have worked under a liaison agreement (memorandum of understanding [MOU]) since 1954. Under the MOU, FTC has primary responsibility for regulating food advertising and FDA for regulating food labeling.60 Recently, FDA and FTC have coordinated efforts in removing fraudulent products from the marketplace, particularly those sold on the Internet and promoted with unproven claims for treating or curing a range of diseases, including diabetes and cancer.
To protect consumers from deceptive claims for dietary supplements, FTC works closely with FDA in employing a 3-pronged strategy that consists of: (1) law enforcement, (2) consumer education, and (3) business outreach. FTC’s authority stems from Section 5 of the FTC Act, which authorizes FTC to prohibit “unfair methods of competition” and “unfair or deceptive acts or practices” in or affecting commerce.61 Section 12 of the FTC Act expressly prohibits “false advertisements” for foods, drugs, medical devices, and cosmetics that are likely to induce consumer purchases.62 Dietary supplement advertising is regulated in a manner consistent with FTC’s regulation of advertising generally under Sections 5 and 12 of the FTC Act. Under FTC law, an advertiser must possess “a reasonable basis” of substantiation to support objective advertising claims, both express and implied, at the time the claim is made. Claims concerning health-related benefits of dietary supplements must be supported by “competent and reliable scientific evidence,” as interpreted by FTC.‡‡‡‡
FTC has extensive investigative and law enforcement authority to prevent companies from engaging in unfair or deceptive acts or practices in dietary supplement advertising, including the dissemination of false ads and unsubstantiated claims. In exercising its consumer protection enforcement authority, FTC may do the following:
Request documents and information from a company through the use of voluntary “access letters;”
Issue subpoenas and “civil investigative demands” requiring production of documentary evidence for any matter under investigation;
Seek and impose consent orders (through administrative or judicial proceedings in federal court);
Seek and obtain consumer redress (e.g., disgorgement of profits, refunds);
Seek and obtain temporary and permanent injunctions, which may include corrective advertising;
Seek and obtain civil penalties;
Seek criminal penalties (by referral to the Justice Department).
FTC administrative and judicial proceedings may be directed against companies and individuals. Responsible principals of a company are typically targeted by FTC in its investigation phase. Endorsers, celebrities, and spokespeople engaged in the promotion of dietary supplements may also be held liable for violations of the FTC Act.
FTC has been active in its enforcement of legal dietary supplement advertising claims. Two years after DSHEA’s passage, in 1996, the Commission issued a resolution authorizing increased FTC investigation of advertisers and marketers of dietary supplements. The resolution provided FTC staff lawyers in any US city the ability to access a company’s substantiating data through a “civil investigative demand” without filing a formal complaint. FTC has continued to exercise an active presence in dietary supplement advertising enforcement. In 1998, FTC issued advertising guidance to the dietary supplement industry, clarifying long-standing FTC regulatory policies with respect to the substantiation of advertising claims, including that consumer testimonials alone may not be used to substantiate claims.63 By that same year, 4 years after passage of DSHEA, FTC had initiated 24 separate dietary supplement enforcement proceedings. In contrast, between 1984 and 1994 (the 10 years preceding DSHEA’s enactment), only 35 cases were initiated by FTC. Between December 2002 and early July 2003, as reported by FTC, the Commission filed or settled 17 enforcement actions against parties engaged in deceptive dietary supplement advertising practices, with consumer sales of the targeted products estimated at more than $1 billion.64 More recently, in May 2010, FTC presented testimony on enforcement activities relating to “Deceptive Marketing of Dietary Supplements” to the Senate’s Special Committee on Aging, stating that during the past decade (2000–2010), the Commission “has filed well over 100 law enforcement actions challenging claims about the efficacy or safety of a wide variety of supplements.”65 According to FTC, the Commission’s law enforcement actions are focused on national advertising campaigns for products with unproven benefits, products promoted to treat or cure serious diseases, products that may present significant safety concerns to consumers, and products that are deceptively marketed to vulnerable population groups such as children or the elderly.9
Indeed, the dietary supplement industry continues to be the focus of increased FTC regulatory scrutiny and enforcement, with significant multi-million dollar civil penalties, redress orders, and other remedies—such as disgorgement of profits—being sought by FTC through federal district courts. A review of recent FTC consent orders provides useful guidance for the general dietary supplement industry. While the substantiation standard continues to be applied on a case-by-case basis, for certain types of health-related claims in dietary supplement advertising, 2 adequate and well-controlled, product-specific clinical trials may be required. Also, when a company relies on third-party ingredient science to substantiate a health-related claim on a finished dietary supplement that has not been clinically tested, the burden will be on the company to demonstrate that the studies were conducted on an “essentially equivalent product.”66 In addition, in a policy shift, FTC has recently made clear that if an advertising claim for a dietary supplement is not permitted under FDA regulations and the governing statute (e.g., the claim indicates disease treatment or prevention, or is not an FDA authorized health claim pursuant to the NLEA), the claim will not be permitted under the FTC Act.67
Similar to FDA, FTC has supplemented its law enforcement actions with business outreach and consumer education programs. FTC has communicated to stakeholders that more robust industry self-regulatory efforts are needed to combat the problem of deceptive health-related claims for dietary supplements, particularly claims relating to weight loss. In 2006, CRN announced the launch and funding of a dietary supplement advertising review program, conducted through the National Advertising Division (NAD) of the Better Business Bureau. In 2009, CRN announced that the NAD dietary supplement review program would be extended for an additional 5 years. The NAD’s mission is to support effective self-regulation of national advertising claims to ensure truth-in-advertising. While the process is voluntary, companies that refuse to participate do so at their own peril. NAD refers such cases to FTC, which gives NAD referrals priority. CRN’s commitment to the self-regulatory process and funding support ($1.5 million over 8 years) has allowed the NAD to hire a dedicated staff attorney to focus solely on dietary supplement advertising claims. During the first 3 years of the program, the NAD opened more than 75 dietary supplement advertising cases, compared to fewer than 10 cases from the year before the CRN/NAD initiative began. At NAD’s annual legal conference in 2008, FTC Commissioner J. Thomas Rosch praised the CRN/NAD initiative as “an excellent example of self-regulation that will increase monitoring of advertising for dietary supplements” and encouraged companies to file a competitive challenge with NAD “if they see a supplement ad that’s misleading, untruthful, or includes claims that can’t be substantiated.”68 §§§§
Role of Other Regulatory Agencies
Other regulatory agencies collaborate with FDA to assist the agency in its mission to protect and advance public health, such as the Drug Enforcement Administration (DEA) and US Customs and Border Protection (CBP), among other agencies. While DEA does not have statutory authority to enforce DSHEA (or the FDCA), FDA has statutory authority to investigate the illegal manufacture and distribution of anabolic steroids included in or sold as dietary supplements under the Controlled Substances Act.69 DEA continues to investigate and uncover products wrongfully labeled as dietary supplements that contain either controlled anabolic steroids or designer steroids that are structurally similar to testosterone. Also, pursuant to the FDA Food Safety Modernization Act of 2011, FDA is required to notify DEA if the agency determines that information submitted in a new dietary ingredient notification is inadequate to establish the safety of the ingredient because it is an anabolic steroid or analogue. The notification to DEA must include the name of the dietary supplement and the name of the person(s) who submitted the notification and/or market the product.70
FDA has broad authority to examine, hold, detain, and prevent the importation of dietary supplements (and other FDA-regulated products) under section 801 of the FDCA.71 FDA is assisted in its mission to prevent the importation of unsafe and improperly labeled dietary supplements by CBP, which is within the Department of Homeland Security. FDA and CBP have had a long history of close cooperation, which was strengthened in 2003 when FDA and CBP signed a MOU. Under the MOU, FDA is allowed to commission thousands of CBP officers in ports and other locations to conduct, on FDA’s behalf, investigations and examinations of imported foods. The agreement significantly strengthened FDA’s ability to implement the Public Health Security and Bioterrorism Preparedness and Response Act of 2002 (BioTerrorism Act).72
FDA Collaborations with Non-Regulatory Governmental Organizations
As part of the Department of Health and Human Services, the CDC is charged with monitoring, detecting, and investigating health problems and conducting research to enhance the protection of public health. When ephedra was still on the market as a dietary supplement, CDC contributed to identifying adverse event reports by coordinating this effort with numerous state Departments of Health.73 In addition, CDC manages surveillance studies, such as the National Health and Nutrition Examination Survey (NHANES), which is a program of studies designed to assess the health and nutritional status of adults and children in the United States, including usage patterns of dietary supplements.74
Conclusion
The years since the enactment of DSHEA have been marked by a progressive development of regulations and guidance by FDA to help ensure that dietary supplements are safe, of high quality, and appropriately labeled. Not surprisingly, there has not been an absolute consensus in the appropriateness or reasonableness of FDA’s approach to regulating dietary supplements. Nonetheless, the core elements of the regulatory infrastructure envisioned by the authors of DSHEA are in place for dietary supplements. These elements are common to every major industry regulated by FDA, and include the following: legal definition for the category; a committed leadership within the agency; a comprehensive claims structure designed to ensure consumers have access to truthful and non-misleading information about a product’s health benefits; standardized labeling requirements (e.g., statement of identity, Supplement Facts panel, list of ingredients) for all products in the category; a defined safety standard; a post-marketing surveillance system; strong legal authority for the agency to remove products with unacceptable health/safety risk; health professional and consumer outreach for product safety alerts; and a standard of manufacturing (i.e., GMPs) to help ensure quality products. With these elements, the $27 billion dietary supplements industry clearly represents a major FDA regulated industry.75 With the complex, interrelated infrastructure of regulations now in place to implement DSHEA and subsequent amendments, the myth that dietary supplements are an unregulated industry is definitely dispelled.
This article was peer reviewed by numerous food and drug law attorneys with extensive expertise in the dietary supplements industry.
R. William Soller, PhD
Dr. Soller is executive director of the Center for Consumer Self Care Health Sciences and Clinical Professor of Pharmacy in the School of Pharmacy at the University of California, San Francisco (USCF). Dr. Soller has broad-based experience in drug and food law, regulations and health policy, and in consumer and pharmaceutical care research at the national and international levels. He is principal author of numerous drug and dietary supplement-related submissions to the FDA on a wide range of subjects related to self-medication. Dr. Soller received his PhD in Medical Sciences from Cornell University Graduate School of Medical Sciences (Neurobiology & Behavior/Pharmacology), and worked in the pharmaceutical and dietary supplement industries for over 20 years before moving to UCSF in 2002, where currently he teaches health policy, manages a telepharmacy service, conducts research, and consults with industry.
Holly J. Bayne
Holly Bayne is a Washington, DC-based attorney and founder of The Law Office of Holly Bayne, P.C., which specializes in the practice of food and drug law. Ms. Bayne focuses on matters relating to the regulation of functional foods, dietary supplements, and botanicals by the FDA, FTC, and other regulatory agencies. She began her legal career at Hyman, Phelps & McNamara, P.C., the largest law firm in the U.S. exclusively devoted to the practice of food and drug law. Before law school, Ms. Bayne worked as an educator and marketing executive in the dietary supplement and cosmetic industries. She has spoken domestically and internationally on issues concerning the regulation of food, dietary supplement, and botanical products, and has written numerous papers and articles pertaining to these issues. Ms. Bayne is admitted to practice law in the District of Columbia and California, and is also a member of the American Bar Association.
Christopher Shaheen
Christopher Shaheen is a researcher in the Center for Self Care at UCSF School of Pharmacy. Shaheen has a research background in basic and social sciences and experience working in pharmaceutical companies and academic research settings. He is a graduate of the University of California, Santa Cruz, and student of the Class of 2015 at UCSF School of Pharmacy.
*Only dietary ingredients are excluded from the food additive provisions of the law; however, other ingredients such as excipients (e.g., preservatives, binders, encapsulation materials, etc.) must be FDA-approved food additives or generally recognized as safe (GRAS) for the intended uses.
†An NDI notification to FDA for an ingredient’s being newly introduced for dietary supplement use is not needed if the ingredient is present in the food supply as an article used for food in a form in which the food has not been chemically altered.
‡A noted exception is the Pearson v. Shalala litigation, holding that FDA’s regulations and enforcement policies governing the use of health claims (under the Nutrition Labeling and Education Act of 1990) on dietary supplements were more extensive than necessary, thus violating the First Amendment. The litigation paved the way for the use of “qualified health claims” in conventional food and dietary supplement labeling. [Pearson v. Shalala, 164 F. 3d 650 (1999).]
§21 U.S.C. § 321(ff). Under DSHEA, a “dietary supplement” is a product intended for ingestion that contains one or more of the following dietary ingredients: a vitamin; mineral; herb or other botanical; amino acid; a dietary substance for use by man to supplement the diet by increasing the total dietary intake; or a concentrate, metabolite, constituent, extract, or combination of any ingredient listed above.
**Federal Food, Drug, and Cosmetic Act; Pub L. No. 75-717, 52 Stat. 1040 (1938), as amended. The FDCA of 1938 also gave FDA the authority to regulate cosmetics and medical devices.
††The drug definition at section 201(g) of the FDCA, “food” articles (or products) intended to affect the structure or any function of the body are exempt from the drug definition. 21 U.S.C. § 321(g)(1)(C).
‡‡For example, in 1985, FDA issued an Import Alert on Evening Primrose Oil (EPO), instructing FDA officials to detain at the border EPO labeled for food use. FDA’s position was that EPO was an unsafe food additive. In 1992, the US Court of Appeals for the Ninth Circuit upheld a lower court’s ruling that EPO was an unsafe “food additive,” preventing the marketing of what is generally considered a safe herbal ingredient. Also in 1992, FDA agents and armed sheriffs raided a Tahoma, Washington, medical clinic seizing bottles of L-tryptophan and samples of injectable B vitamins from Germany used by Jonathan Wright, MD, in his clinic.
§§The major industry trade associations: American Herbal Products Association (AHPA), Consumer Healthcare Products Association (CHPA), Council for Responsible Nutrition (CRN), Natural Products Association (NPA, formerly the National Nutritional Foods Association [NNFA]), United Natural Products Alliance (UNPA, formerly Utah Natural Products Alliance).
***Dr. Jane E. Henney, MD, was FDA Commissioner from January 1999 to January 2001. She started as Commissioner in November 1998.
†††Dr. David A. Kessler, MD, JD, was FDA Commissioner from November 8, 1990 to February 28, 1997. Michael A. Friedman, MD, served as lead deputy commissioner while the post of commissioner was vacant from February 28, 1997 to November 30, 1998.
§§§This is the same definition used for a serious adverse drug experience (or event) (SADE). See: www.fda.gov/newsevents/publichealthfocus/ucm155683.htm. FDA letter to Iovate.
****As defined in the final rule, quality means that the dietary supplement “consistently meets the established specifications for identity, purity, strength, and composition, and has been manufactured, packaged, labeled, and held under conditions to prevent adulteration under section 402(a)(1), (a)(2), (a)(3), and (a)(4) of the Federal Food, Drug and Cosmetic Act.”
††††AHPA has been discussed elsewhere in this article. AHP’s mission is to promote the responsible use of safe and effective herbal products through the development of botanical standards of identity, purity, and analytical methods, as well as to critically review traditional and scientific data regarding their efficacy and safety.
‡‡‡‡“Competent and reliable scientific evidence” has been defined through FTC case law as: “tests, analyses, research, studies, or other evidence based on the expertise of professionals in the relevant area, that have been conducted and evaluated in an objective manner by persons qualified to do so, using procedures generally accepted in the profession to yield accurate and reliable results. See FTC Enforcement Policy Statement on Food Advertising (May 1994) at www.ftc.gov/bcp/policystmt/ad-food.shtm.
§§§§See also a voluntary industry program through the Natural Products Foundation. Truth In Advertising Program. Available at: http://naturalproductsfoundation.org/index.php?src=gendocs&ref=truth_in_advertising&category=FoundationPrograms.
References
- Dietary Supplement Health and Education Act of 1994. Public Law 103-417: 103rd Congress. October 25, 1994. Available at: http://dshedu.com/DSHEA_Legal/dshea.html.
- Soller RW. Regulation in the herb market: the myth of the “unregulated industry.” HerbalGram. 2000:49;64-67.
- 21 U.S.C. § 321
- Food Safety and Modernization Act. Public Law 111-353. Statute 3885. January 4, 2011. Available at: www.gpo.gov/fdsys/pkg/PLAW-111publ353/content-detail.html. Accessed October 12, 2011.
- 21 U.S.C. § 342(a)
- 21 U.S.C. § 343(j).
- Amendment: Public Health Service Act. Public Law 94-278. Staute 401. April 22, 1976.
- Nutrition Labeling and Education Act. Public Law 101-533. 104 Statute 2353. 1990.
- 984 F.2d 814. 61 USLW 2491. US v. Two Plastic Drums, More Or Less Of An Article Of Food, Labeled In Part: Viponte Ltd. Black Currant Oil Batch No. Boosf 039, etc., and Traco Labs, Incorporated. No. 92-1172. United States Court of Appeals, Seventh Circuit. Argued October 21, 1992. Decided January 27, 1993.
- 987 F. 2d 33. US v. 29 Cartons Of *** An Article of Food, Etc., Defendant Oakmont Investment Co., Inc. Appeal From The United States District Court For The District Of Massachusetts. First Circuit. March 3, 1993.
- Shelton S. Executive interview: Q&A with Bob Ullman. Engredea News & Analysis. September 8, 2005. Available at http://newhope360.com/executive-interview-qa-bob-ullman. Accessed October 12, 2011.
- Hutt P. The history & future of the dietary supplement industry. Natural Products Insider. September 21, 2009. Available at: www.naturalproductsinsider.com/articles/2009/09/the-history-future-of-the-dietary-supplement-health-education-act.aspx. Accessed February 24, 2011.
- McNamara S. Dietary supplements of botanicals and other substances: a new era of regulation. Food Drug Law J. 1995;50:341-355.
- 103rd Congress. Second Session. Senate Report 103-410. Dietary Supplement and Health Education Act 1994. October 8, 1994. Report from the Committee on Labor and Human Resources to accompany S.784: p. 17.
- Wertheimer L. FDA issues new standards for dietary supplements. NPR News. June 23, 2007. Available at: www.npr.org/templates/story/story.php?storyId=11326842.
- Landau D. Dietary supplement industry unregulated. Injury Board Blog Network. February 1, 2011. Available at: http://fairfax-loudoun.injuryboard.com/fda-and-prescription-drugs/dietary-supplement-industry-unregulated.aspx?googleid=288118. Accessed February 24, 2011.
- Green A. Spreading the blame: examining the relationship between DSHEA and the baseball steroid scandal. Boston Univ Law Rev. 2010;90:399-430.
- Henney J. Implementation of the Dietary Supplement Health Education Act. Testimony before the House Committee on Government Reform. March 25, 1999. FDA Speech Archives. Available at: www.fda.gov/NewsEvents/Testimony/ucm115082.htm.
- Statement by Jane E. Henney, MD, Commissioner Food and Drug Administration, Department of Health and Human Services, Before the Committee on Government Reform, US House of Representatives. March 25, 1999. FDA Speech Archive. Available at: .
- David A. Kessler, MD—FDLI Annual Meeting. December 13, 1994. FDA Speech Archives. Available at: www.fda.gov/NewsEvents/Speeches/SpeechArchives/ucm054775.htm.
- Federal Register Final Rule: Regulations on statements made for dietary supplements concerning the effect of the product on the structure or function of the body. Federal Register. January 6, 2000;65(4):999-1050.
- Proposed Rule: Regulations on statements made for dietary supplements concerning the effect of the product on the structure or function of the body. Federal Register. April 29, 1998;63(82):23623-23632.
- 21 C.F.R. § 101.14(a)(5)
- FDA dietary supplement strategy (ten-year plan) webpage. Grimes & Reese PLLC website. Available at: www.mlmlaw.com/library/guides/fda/ds-strat.html#Outline. Accessed October 12, 2011.
- Levitt, JA; Regulation of dietary supplements: FDA’s strategic plan. Food and Drug Law J. 2002;57:1.
- Joseph A. Levitt, Esq. (FDA director, CFSAN) on March 20, 2001 before the House Committee on Government Reform Available at: www.fda.gov/NewsEvents/Testimony/ucm115229.htm.
- McClellan M. Ephedrine alkaloid-containing dietary supplements. Statement before the Subcommittees on Commerce, Trade, and Consumer Protection and Oversight and Investigations, House Committee on Energy and Commerce. July 24, 2003. Available at www.fda.gov/NewsEvents/Testimony/ucm115044.htm. Accessed February 24, 2011.
- Crawford L. Speech before Council of Responsible Nutrition Annual Conference. October 25, 2004. Available at: www.fda.gov/NewsEvents/Speeches/ucm053325.htm.
- Final Rule: Declaring dietary supplements containing ephedrine alkaloids adulterated because they present an unreasonable risk. Federal Register. February 11, 2004;69(28);6788-6854.
- 109th Congress (2005-2006)—Senate Report 109-324. Dietary Supplement and Nonprescription Drug Consumer Protection Act.
- 21 U.S.C. § 374(a); FDA authority for factory inspection, to enter at reasonable times any factory, warehouse, or establishment in which food, drugs, devices, tobacco products, or cosmetics are manufactured, processed, packed or held for introduction into interstate commerce.
- FDA Guidance for Industry: Questions and answers regarding adverse event reporting and recordkeeping for dietary supplements as required by the dietary supplement and nonprescription drug consumer protection act; Draft Guidance issued October 2007; revised June 2009. Available at: www.fda.gov/Food/GuidanceComplianceRegulatoryInformation/GuidanceDocuments/DietarySupplements/ucm171383.htm.
- Medical Dictionary for Regulatory Activities Maintenance and Support Services Organization website. Available at: https://meddramsso.com. Accessed October 12, 2011.
- Safety Monitoring of Medicinal Products: Guidelines for Setting Up and Running a Pharmacovigilance Centre. World Health Organization website. Available at: http://apps.who.int/medicinedocs/en/d/Jh2934e/.
- Health Hazard Evaluation Board: Hydroxycut. Available at: www.fda.gov/downloads/NewsEvents/PublicHealthFocus/UCM160672.pdf. Accessed October 12, 2011.
- FDA Letter to Iovate Health Sciences, Inc. April 30, 2009. Available at: www.fda.gov/NewsEvents/PublicHealthFocus/ucm155683.htm.
- 21 U.S.C. § 342 (g); 21 C.F.R. § 110. 3 et seq.
- FDA Advance Notice of Proposed Rule Making: Current good manufacturing practice in manufacturing, packing, or holding dietary supplements. Federal Register. February 6, 1997;62:5700.
- Final Rule: Current Good Manufacturing Practice in Manufacturing, Packaging, Labeling, or Holding Operations for Dietary Supplements. Federal Register. August 24, 2007;72: 34752-34958.
- 21 U.S.C. § 342 (g)(1)
- Personal communication from Brad Williams (CFSAN, FDA) to Mark Blumenthal (American Botanical Council). January 13, 2011.
- Warning Letter to Coats International Holdings, Inc. US Food and Drug Administration. March 30, 2010. Available at: www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm210182.htm.
- 21 U.S.C. § 343(s)(1)(2)(D). Federal Food, Drug, and Cosmetic Act. Misbranding provision.
- AOAC capabilities. AOAC website. Available at: www.aoac.org/about/aoac_capabilities.pdf.
- 21 C.F.R. § 2.19. FDA, Code of Federal Regulations, “Methods of Analysis.”
- Saw Palmetto Dietary Supplment Standard Reference Materials. National Institute of Standards and Technology (NIST) website. Available at: www.nist.gov/mml/analytical/organic/sawpalmetto.cfm.
- Dietary Supplements, Functional Food, and Beverages. NSF International website. Available at: .
- NPA GMP Certification Program Overview. Natural Products Association website. Available at: www.npainfo.org/index.php?src=gendocs&ref=NPAGMPCertificationProgramOverview&category=GMPcert.
- John Taylor, FDA Associate Commissioner for Regulatory Affairs. Statement before the Senate Committee on Commerce, Science and Transportation. October 23, 2003. Available at: www.fda.gov/NewsEvents/Testimony/ucm114985.htm.
- Robert Brackett, Director, Center for Food Safety and Applied Nutrition Dietary Supplement Safety Act: How is FDA Doing 10 Years Later. Statement before the Subcommittee on Oversight of Government Management, the Federal Workforce, and the District of Columbia Senate Committee on Governmental Affairs. June 8, 2004. Available at: www.fda.gov/NewsEvents/Testimony/ucm113767.htm.
- Sharfstein J, FDA Principal Deputy Commissioner. Oversight of Dietary Supplements 2010. Statement before the Special Committee on Aging, U.S. Senate. May 26, 2010. Available at: www.fda.gov/NewsEvents/Testimony/ucm213531.htm.
- Dietary supplement alerts and safety information. US Food and Drug Administration website. Available at: www.fda.gov/Food/DietarySupplements/Alerts/default.htm. Accessed February 24, 2011.
- OTC retail sales—1964-2009. Consumer Healthcare Products Association website. Available at www.chpa-info.org/pressroom/Retail_Sales.aspx. Accessed February 24, 2011.
- The medical device market: USA. Espicom website. Available at: www.espicom.com/Prodcat2.nsf/Product_ID_Lookup/00000110?OpenDocument. Accessed February 24, 2011.
- IMS Health reports U.S. prescription sales grew 5.1 percent in 2009, to $300.3 billion [press release]. Norwalk, CT: IMS Health. Available at: www.imshealth.com/portal/site/imshealth/menuitem.a46c6d4df3db4b3d88f611019418c22a/?vgnextoid=d690a27e9d5b7210VgnVCM100000ed152ca2RCRD. Accessed February 24, 2011.
- U.S. food industry overview. Plunkett Research Inc website. Available at: www.plunkettresearch.com/food%20beverage%20grocery%20market%20research/industry%20statistics. Accessed February 24, 2011.
- FDA transparency initiative. US Food and Drug Administration website. Available at: www.fda.gov/AboutFDA/Transparency/TransparencytoRegulatedIndustry/PhaseIIITransparencyReport/FDATransparencyInitiative/default.htm. Accessed October 12, 2011.
- FDA Commissioner Hamburg Letter to Manufacturers of Dietary Supplements on Undeclared Active Drug Ingredients. December 15, 2010. Available at: www.fda.gov/downloads/Drugs/ResourcesForYou/Consumers/BuyingUsingMedicineSafely/MedicationHealthFraud/UCM236985.pdf
- United States v. Park 421 U.S. 658-672. 1975.
- Comments of the Staff of the Bureau of Consumer Protection of the Federal Trade Commission: In the Matter of Regulations on Statements Made for Dietary Supplements Concerning the Effect of the Product on the Structure or Function of the Body; Proposed Rule August 27, 1998. Available at www.ftc.gov/be/v980023.shtm#N_1_
- 15 U.S.C. § 45. Federal Trade Commission Act.
- 15 U.S.C. § 52. Federal Trade Commission Act.
- FTC, Dietary Supplements: An Advertising Guide for Industry, p. 9 (Nov. 18, 1998).
- Federal Trade Commission Attacks $1 Billion In Deceptive Health Marketing since December [press release]. Washington, DC: Federal Trade Commission. Available at: www.ftc.gov/opa/2003/07/diethealth.htm. Accessed October 12, 2011.
- Prepared Statement of the Federal Trade Commission on “Deceptive Marketing of Dietary Supplements FTC Enforcement Activities” Before the Special Committee on Aging, United Sates Senate, Washington, DC. May 26, 2010. Available at: www.ftc.gov/os/testimony/100526dietarysupplementstatement.pdf.
- Federal Trade Commission Decisions, Findings, Opinions, and Orders, January 1, 2011 to June 30, 2011: Volume 151. Available at: www.ftc.gov/os/decisions/docs/volume151.pdf
- Iovate Health Sicences USA, FTC Case No. 10-cv-587(WDNY July 29, 2010). Available at: www.ftc.gov/os/caselist/0723187/100729iovatestip.pdf.
- CRN foundation announces five-year grant to national advertising division for review of supplement ads [press release]. Washington, DC: Council for Responsible Nutrition. Avilable at: www.crnusa.org/CRNPR09CRNFoundationAnnouncesFiveYearGranttoNAD110909.html. Accessed October 12, 2011.
- Anabolic Steroid Control Act of 2004; 21 U.S.C. § 802(41)(A).
- 21 U.S.C. § 350b(c)(1)(2).
- 21 U.S.C. § 381; FDA’s authority under the FDCA to regulate imports and exports.
- The Public Health Security and Bioterrorism Preparedness and Response Act of 2002. Pub. Law No. 107-188; 116 Stat 594. 107th Congress.
- Centers for Disease Control and Prevention. Adverse Events Associated with Ephedrine-Containing Products—Texas, December 1993–September 1995. MMWR. 1996;45:689-693.
- National Health and Nutrition Examination Survey. Centers for Disease Control and Prevention website. Available at: www.cdc.gov/nchs/nhanes.htm.
- 2010 Supplement Business Report. Nutrition Business Journal.
|
