Issue:
99
Page: 66-69
Botanical New Drug Applications – The Final Frontier
by Freddie Ann Hoffman, MD, Steven R. Kishter, MD, DDS
HerbalGram.
2013; American Botanical Council
Marketing botanical ingredients as foods and dietary ingredients in the United States is
commonplace. Getting them approved as prescription drugs is a somewhat new
frontier, and so far only two botanicals have achieved this goal. On October
31, 2006, the US Food and Drug Administration (FDA) approved the first
botanical drug, Veregen® (sinecatechins; ointment, 15%; Medigene,
Planegg/Martinsried, Germany), a proprietary extract of green tea (Camellia sinensis Kuntze, Theaceae)* for
treating genital and perianal warts.1 FDA approved the second
botanical New Drug Application (NDA) on December 31, 2012, for FulyzaqTM
(crofelemer; 125 mg tablet; Salix Pharmaceuticals; Raleigh, North Carolina),
the first oral prescription botanical drug for the novel indication of
HIV-associated diarrhea. Fulyzaq is a proprietary extract of the blood-red
latex of the South American croton tree (Croton
lechleri Müll Arg, Euphorbiaceae).2,3
Defining
“Botanical Drug”
FDA defines a
“botanical” as a finished product containing ingredients and/or constituents of
vegetable matter. This classification includes whole plants or plant parts —
including plant materials such as juices, gums, fatty oils, scent oils, etc. —
and also includes algae or macroscopic fungi and similar products. Excluded are
fermentation products, isolated and purified ingredients, or homeopathic
ingredients, all of which already have well-described drug regulatory pathways
in the United States.4
Because both
Fulyzaq and Veregen are intended to diagnose, treat, prevent, mitigate, or cure
an abnormal condition, they are considered “drugs.” In particular, they are
“new” drugs, i.e., drugs marketed in the United States after 1938 that prior to
approval were “not generally recognized as safe and effective under the
conditions prescribed, recommended, or suggested in the labeling.”5 As new drugs, the
sponsors were required to submit NDAs for FDA pre-market approval. Each product
underwent extensive product and clinical development to meet the drug
requirements and to document safety and efficacy for its intended use(s).
Unlike foods, dietary supplements, or cosmetic products, which are restricted
from making disease claims, these botanical drugs can make disease claims that are
supported by their approved NDAs. Both products are available only by
prescription.
Initial Steps:
Investigational New Drug (IND) Application
Botanical drug
development begins long before FDA’s review process. A botanical must undergo
identification and taxonomic classification. Raw material sourcing as well as
collection, manufacturing, and formulation practices must be described
adequately. Details of any prior and current human use also are important to
obtain, as such information can significantly impact the regulatory
requirements.4
After these
critical steps, filing an Investigational New Drug (IND) application with FDA
is the first part of the formal drug regulatory process that culminates with
FDA’s decision on the NDA. An IND exempts the “investigational new drug” from
federal requirements that it must be safe
and effective, allowing for research
and development activities to take place within US borders.6
Following an
initial submission, FDA has 30 calendar days to review the IND. For early (Phase
1) clinical development, FDA focuses on the drug’s safety, which is based on
its chemistry, toxicity profile, and use history. Novel chemicals submitted
under IND require extensive nonclinical toxicology work, even for Phase 1. A
botanical drug with a well-documented history of human use can often circumvent
much of the toxicology requirements, at least initially, although some safety
testing is generally required.4
Ultimate Step:
New Drug Application (NDA)
Because new
drugs must undergo FDA pre-market approval, NDA submission is the ultimate step
in the development process.7 The NDA is a collation of data and
analyses collected under the IND, a summary of which will become the drug
package insert to support drug labeling and promotion. Requesting a “pre-NDA”
meeting with FDA helps drug sponsors ascertain whether FDA agrees with their
marketing proposals. FDA expects to meet with the NDA sponsor to discuss the
content and format of the application prior to its submission, as well as any
unresolved issues raised during the IND, and any further requirements for
potential approval. This may include completion of nonclinical testing, pivotal
trials analyses, and submission of key clinical study databases for FDA’s own
review and analysis. The sponsor also will need to submit the status of any
unexpired patents for the drug.8 FDA even has final say over the
drug’s brand and scientific (“generic”) names.9
The most
important step of the process and, for botanicals, the most difficult step to
satisfy, is FDA’s review and acceptance of the chemistry, manufacturing, and
controls (CMC). At the initial development stages (Phase 1 and early Phase 2),
CMC requirements are relaxed. For example, FDA often allows the current nondrug
formulation to be used in early phase studies. To support the NDA, however,
later-stage (i.e., Phase 2b and
pivotal†) clinical studies must be conducted using finished drug
product that conforms to pharmaceutical requirements (Good Manufacturing
Practices). FDA also will want to review and negotiate the sponsor’s plans for
commercialization, including manufacturing scale-up, packaging, and lot-release
protocols to be utilized in the commercial production of the drug following
approval. Finally, the sponsor must be prepared for FDA to conduct a
pre-approval inspection of the manufacturing facilities.
NDA Approval
Requirements
FDA can approve
the NDA when the drug meets the legal requirements discussed herein. Federal
law requires that a new drug be safe and effective for its intended use, as
demonstrated by substantial evidence,
defined as “evidence consisting of adequate and well-controlled investigations,
including clinical investigations, by experts qualified by scientific training
and experience to evaluate the effectiveness of the drug involved.”
Additionally, data from the substantial evidence demonstration must show that
the “drug will have the effect it purports or is represented to have under the
conditions of use prescribed, recommended, or suggested in the labeling….”5
Control over the drug’s lot-to-lot variation also must be adequately addressed.7
Botanical drug
approval is a very different process from acceptance of the same ingredient as
a non-drug. For example, foods and dietary supplements are allowed to be
marketed if they are food ingredients or ingredients that are “generally
recognized as safe” (GRAS), or contain ingredients with “a history of use or
other evidence of safety” that “will
reasonably be expected to be safe…”5 In contrast, a drug must
demonstrate that its benefits outweigh its risks to the population for which
its use is intended.
NDA Review
Timeline and the “Prescription Drug User Fee Act” (PDUFA)
FDA’s timelines
for NDA reviews are guided by the current Prescription Drug User Fee Act
(PDUFA). Originally enacted in 1992 by the US Congress to reduce lengthy NDA
review times, PDUFA authorizes FDA to collect fees from sponsors whose
applications require Agency review. PDUFA fees support the Agency’s review
processes by allowing the hiring of experts and other activities. No fees are
required if the NDA sponsor and/or product fits one or more of the following
scenarios: first-time filer, product for a rare disease or condition under an
Orphan Product Designation, product deemed necessary to protect the US public health,
product for which user fees will pose a significant barrier to innovation due
to limited resources or other circumstances, or a sponsor that is a small
business (<500 employees) that does not yet have an approved drug on the US
market.10
Upon receipt of
an NDA, FDA has 60 days to review and accept it for filing. Incomplete or
poorly organized applications can result in a “failure to file” notice. FDA’s
review “clock” does not start until the Agency allows the NDA to be filed
(referred to as the “acceptance to file” notification; see Figure 1). Once the
NDA is filed, the Agency sets a goal (PDUFA) date — the date by which FDA
should act on the application (see Table 1
on page 68). 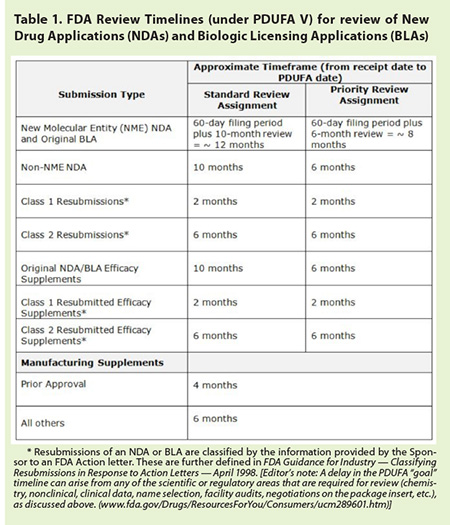
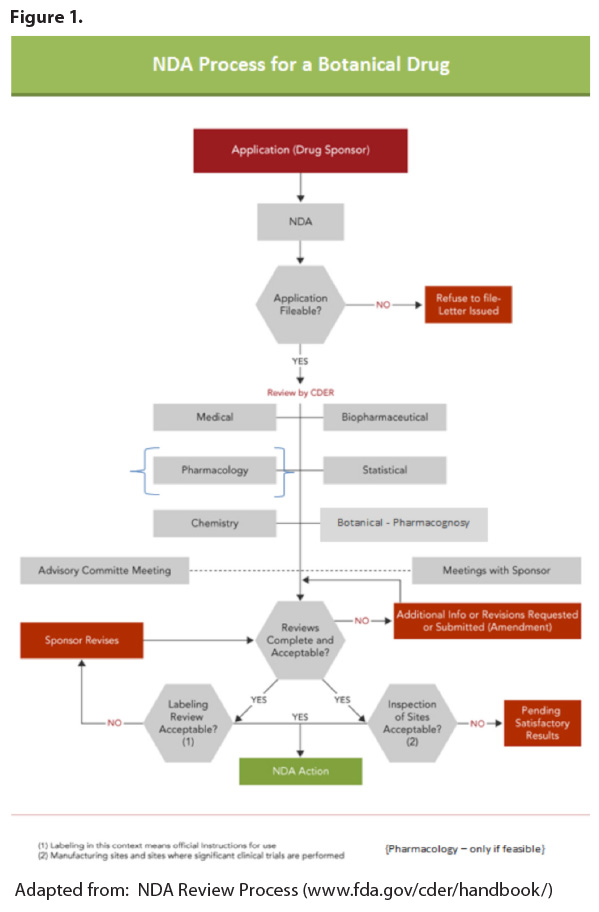
FDA can modify
the review timeline. It can hold the clock while waiting for a response to its
request for information. Or, the timeline can be condensed when FDA assigns
“Priority Review” to NDAs for drugs intended to treat a serious and
life-threatening condition lacking satisfactory treatments. FDA also can
utilize an “Accelerated Approval” process to decide on a drug prior to
receiving all safety or efficacy data needed for approval. Although a drug can
be marketed following Accelerated Approval, its sponsor will be required to
collect and to submit additional efficacy and safety data at a later time for
FDA to determine if the drug should remain on the market. If these data do not
continue to support the drug’s safety or efficacy, FDA can revoke the approval.11
On July 9, 2012,
President Obama signed into law the fifth reauthorization of the Prescription
Drug User Fee Act. Known as “PDUFA V,” the new law took effect on October
1, 2012. It includes the following timeframes used by FDA to project a calendar
day (called the “PDUFA date”) — a goal, but not
a deadline — by which it plans to return a decision, following the Agency’s
review of the various types of marketing applications.
Botanical Drug
Approvals
More than 500
pre-IND meetings and IND applications have been submitted to FDA for botanical
drugs; two botanical drug NDAs have been submitted to FDA and both were
approved. While it appears that many sponsors have accomplished the IND step,
only two have reached the final NDA step. Why are there only two FDA-approved
NDAs? Based on these authors’ experience, the following represent three of the
most common reasons that could explain why more botanical drug NDAs have not
been submitted to FDA for review:
Failure to show efficacy: Failure to show
clinically relevant and statistically significant efficacy is the single most
common reason why most drugs — not just botanical drugs — fail to reach the NDA
step. Although many sponsors “believe” that their product “works,” the
stringent criteria for US drug approval consist of documented safety and
efficacy from one or more multicenter “adequate and well-controlled” clinical
studies. For pivotal studies (those efficacy studies that will be used to
support the NDA), it is very important that target populations be
well-circumscribed by the protocol eligibility criteria, with appropriate and
FDA agreed-upon outcome measures, proper controls (e.g., placebo or active treatment), and be well-monitored and
accurately analyzed.
Unrealistic Expectations: Inexperienced
drug sponsors often have unrealistic expectations when it comes to planning and
executing a drug development program. This may be due, in part, to FDA’s
relatively relaxed requirements during initial stages of IND development, which
may give sponsors a false sense of security that the requirements for
botanicals are less rigorous than those for non-botanical drugs. It also may be
due to the fact that regulatory requirements for botanicals are not
internationally harmonized, as they are for other drug categories, which
creates confusion, because US requirements differ from those of other
countries. Also, many botanical drug sponsors have never developed a drug for
the US market, or come from different industries or regulatory environments.
Some sponsors are unwilling to accept — or simply deny — that the United States
requires submission of “raw” data (chemistry, nonclinical safety testing,
clinical study databases, etc.) to support drug filings, rather than data
summaries or “expert” opinion, as is commonplace in other countries.
Insufficient Funding: Lack of or
insufficient funding to complete the development process is not an uncommon
problem for many botanical drugs under IND. This may be due to the economic
climate, lack of acceptance by the investment community, lack of patent status
(although the product may enjoy other forms of intellectual property that may
be superior to patents), or insufficient planning. Again, many botanical drug
sponsors, particularly those whose products are in other market channels (e.g., dietary supplements) or foreign
markets, underestimate the level of documentation and data that FDA requires to
assess that a drug does what it claims to do in its labeling.
Conclusion
For many
botanical drugs, the path to NDA approval has the potential to be shorter and
less costly than for “standard” new chemical entities. However, until the
botanical community comes to grips with the realities of the legal requirements
for drug approval in the United States, there will continue to be few sponsors
that are able to traverse this final frontier.
*HerbalGram does not customarily refer to
herbs by binomial and authority, but by the Latin binomial and family name
listed in the American Herbal Products Association’s Herbs Of Commerce, 2nd edition. In this instance, the
authors preferred to note the authority in the scientific name because for a
drug, FDA insists on both the Latin bionomial and authority.
†The term pivotal study refers to an adequate
and well-controlled clinical trial designed to evaluate the specific dose,
route, schedule, formulation, and specific clinical indication that will become
the subject of the NDA. In particular, the drug product used for a pivotal
study should meet FDA requirements for commercial manufacturing (Good
Manufacturing Practices or GMPs).
References
- FDA approves special green tea extract as a new topical drug for genital warts [press release]. Austin, TX: American Botanical Council; November 9, 2006. Available at: http://cms.herbalgram.org/press/gthpv.html. Accessed April 30, 2013.
- FDA approves first anti-diarrheal drug for HIV/AIDS patients [press release]. Silver Spring, MD: US Food and Drug Administration; December 31, 2012. Available at: www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm333701.htm. Accessed April 30, 2013.
- FDA approves crofelemer as first-ever oral botanical drug [press release]. Austin, TX: American Botanical Council; January 2, 2013. Available at: http://cms.herbalgram.org/press/2013/FDA_Approves_Crofelemer.html. Accessed April 30, 2013.
- Guidance for Industry on Botanical Drugs Products. US Department of Health and Human Services. US Food and Drug Administration, Center for Drug Evaluation and Research. June 2004. Available at: www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070491.pdf. Accessed April 30, 2013.
- Federal Food, Drug and Cosmetic Act and its Amendments, 21 USC Chapter 9 §§ 301–399f.
- Investigational New Drug application. US Food and Drug Administration website. Available at: www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/InvestigationalNewDrugINDApplication/default.htm. Accessed April 30, 2013.
- New Drug Application. US Food and Drug Administration website. Available at: www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/NewDrugApplicationNDA/default.htm. Accessed April 30, 2013.
- Applications For FDA Approval To Market A New Drug. Title 21 CFR §314.50.
- Guidance for Industry: Contents of a Complete Submission for the Evaluation of Proprietary Names. US Department of Health and Human Services. Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). February 2010. Available at: www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm075068.pdf. Accessed April 30, 2013.
- Prescription Drug User Fee Act (PDUFA). US Food and Drug Administration website. Available at: www.fda.gov/ForIndustry/UserFees/PrescriptionDrugUserFee/default.htm. Accessed April 30, 2013.
- FDA Commissioner removes breast cancer indication from Avastin label [news release]. Silver Spring, MD: US Food and Drug Administration; November 18, 2011. Available at: www.fda.gov/NewsEvents/Newsroom/ucm279485.htm. Accessed April 30, 2013.
|